Reveal the Functional and Pathological Importance of Architectural Features of the Genome
The 3D structure of the genome is known to have significant impacts on human health and disease. 3D organizational features including loops, TADs, and compartments can modify promoter-enhancer interactions, gene splicing during transcription, gene expression, and DNA replication.
The pathological implications of these alterations include chromosome translocations, the creation of fusion transcripts, and altered regulation of distant genes caused by long-range effects of single nucleotide variants. Capturing 3D genomic information enables more complete characterization of disease mechanisms and identification and validation of novel therapeutic targets and pathways.
Why Choose Arima Technology for Your Research?
Proven performance for greater accuracy of chromatin loop and topological domain detection, even at reduced sequencing depths
Fast and user-friendly workflows to go from sample to discovery using either genome-wide or targeted approaches
Quality you can trust, with built-in QC steps to ensure you get reliable sequencing results every time
Support for a broad range of sample types and an optimized protocol for low sample input
Discover What’s Possible with 3D Genomics

Down-syndrome-induced senescence disrupts the nuclear architecture of neural progenitors.
Explore Disease Mechanisms
The 3D conformation of DNA in a cell has broad impacts on human health and disease — including in neurodevelopmental disorders caused by chromosomal abnormalities. Learn how 3D genomics can be used to understand transcription disruption and its impact on therapeutic interventions.
Discover Genetic Risk Variants
GWAS studies have identified a large number of single nucleotide polymorphisms that are associated with complex disease risk. Learn how 3D chromatin structure can be used to identify disease-causing genes and understand their functional importance.
Watch the webinar to learn about the utility of 3D genomics for understanding disease-associated variants in non-coding regions in autoimmune disorders.
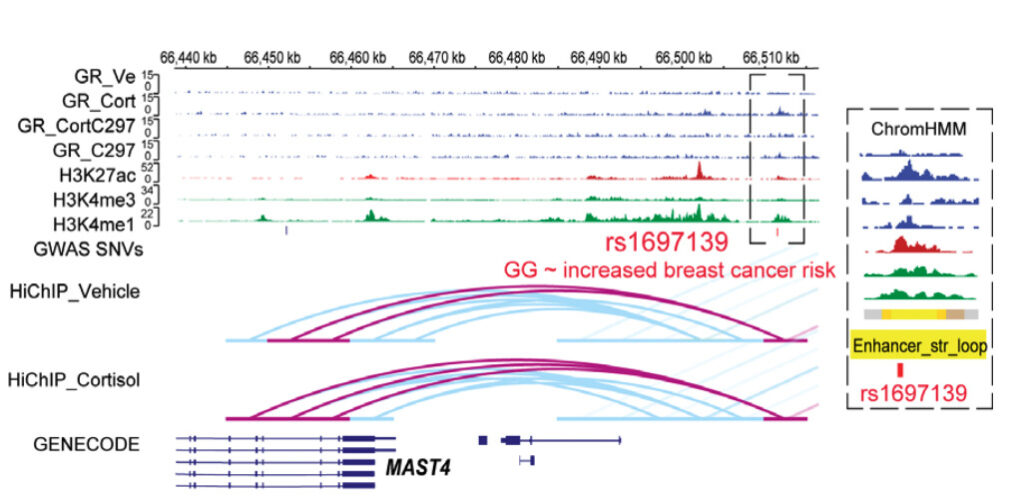
Example of glucocorticoid receptor-dependent pharmacogenomic (PGx)-eQTLs with functional implications for disease risk.
Detect Non-Coding Variants
Understanding non-coding genetic variants and their functional role in human disease remain a significant challenge. Learn how 3D genomics can be used as part of a multi-omic approach to elucidate the role of non-coding variants in the expression of genes involved in autoimmunity, metabolic and mood disorders, osteoporosis, and cancer.
Blog: Unmasking Silent Non-Coding Genetic Risk Variants in Disease with 3D Genomics
Two Ways To Get Started with 3D Genomics
Arima Kits

Use our sample and library preparation kits to advance your understanding of human health and disease
Arima Services

Let our scientists share their expertise in sample prep, library construction, and bioinformatics


