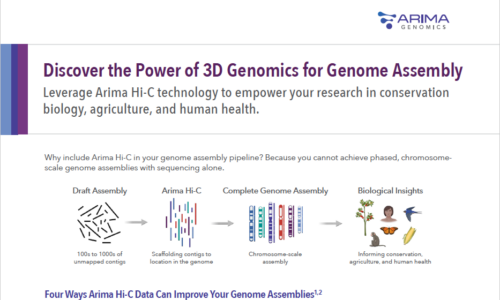
Arima Hi-C: Because You Can’t Get Phased, Chromosome-Scale Genome Assemblies with Sequencing Alone
Great genome biology is built on a foundation of high-quality, phased, chromosome-scale genome assemblies. DNA sequencing technologies have made big strides in achieving long, accurate contigs, but they aren’t enough to deliver platinum quality genomes to support research for years to come. Adding Arima Hi-C data takes your assembly from a draft to a complete reference genome.

Why Choose Arima Hi-C for Your Genome Assembly?
Publication Highlights
From Sample to Chromosome-Scale Genome Assembly
Hi-C Prep
Easy to follow, rapid 6-hour protocol
Library Prep
Use Arima Library Prep Module for standard or low input samples
Sequencing
200 million paired-end reads per Gb of genome
Data Analysis
Assemble and scaffold with tools of your choice
Two Ways To Get Started
Genome Assembly Kits

Use our sample and library preparation kits to bring chromosome-scale assemblies to your lab
Genome Assembly Services

Let our scientists share their expertise in sample prep, library construction, and bioinformatics
Michael Quail
Principal Scientific Manager of DNA Pipelines, Wellcome Sanger Institute“The newer Arima kits are being used because we found them to be robust across a range of tissue types from diverse vertebrate and invertebrate species. Kit robustness to a diversity of samples is an essential requirement for the Darwin Tree of Life Project.”
Gene Myers, PhD
G10K Council Member and Director at Max-Planck Institute of Molecular Cell Biology and Genetics, DresdenExplore Our Genome Assembly Resources
Learn more about some of the hundreds of genomes assembled using Arima Hi-C.