September 19, 2022
Share
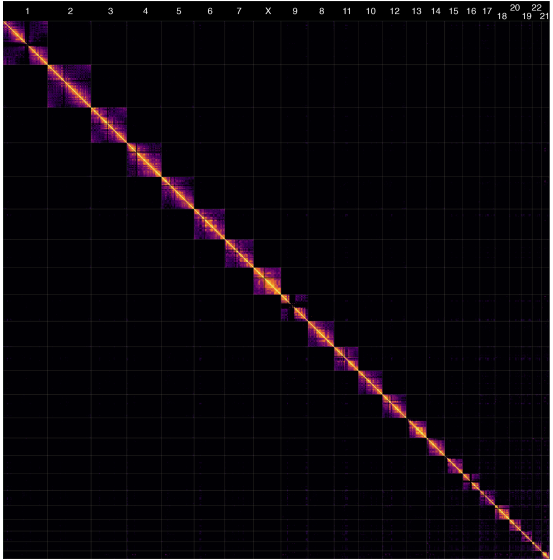
Hi-C heatmap of the first complete human genome. Nurk, et al. 2022.
At the heart of using Hi-C data for genome assembly is probability statistics. It turns out there is a tendency for portions of the genome that are nearby in linear sequence space to also be in close proximity to each other in 3D chromatin space compared to sections of the genome that are farther apart in linear space or on different chromosomes. This enables tools to take advantage of these statistical probabilities to infer information and aid in assembling, scaffolding, and phasing genomes.
While this certainly isn’t a comprehensive list, we’ve collected and organized a catalog of software tools that utilize Hi-C data so that you can leverage these bioinformatics workflows for the goals of your genome project.
1. QC, Contact Maps, and Visualization
By far the most common tool encountered when browsing genome papers that utilize Hi-C data is the Juicer Tools and Juicebox software. This set of tools processes Hi-C data and outputs contact maps that have structural features annotated. Originally published in 2016 by Durand, et al., the tools have consistently been updated and maintained as Hi-C data has become more ubiquitous via the GitHub repository.
HiGlass, a web-based tool for exploring genome interaction maps, enables navigation of 2D genomic maps alongside 1D genomic tracks. Originally published in 2018 by Kerpedjiev, et al., this tool dynamically arranges views of several Hi-C datasets to support multiscale contact maps and genomic data track visualization across multiple resolutions, loci, and conditions.
Another suite of tools that include visualization is HiCExplorer, which categorizes itself a set of programs to process, normalize, analyze, and visualize Hi-C data. Originally published in 2018 by Wolff et al., it is intended for users with little bioinformatic background to perform every step in the needed analysis in one workflow.
As part of a suite of tools developed by the High Performance Assembly Group at the Wellcome Sanger Institute, PretextView, is a desktop application for viewing pretext contact maps to aid in detecting scaffolding issues as part of a broader curation strategy outlined in Howe, et al 2021.
2. Ordering, Orienting, and Fixing Mis-assemblies
Of course, once you have checked the quality and visualized the data, the next important step is to apply the information in the Hi-C data to improve a draft genome assembly. There’s no shortage of open source tools for de novo genome scaffolding, but here’s a few of the most used ones we’ve encountered.
3D-DNA, originally published in 2017 by Dudchenko, et al., is a pipeline used to address misjoins, anchor, order, and orient the contigs of a draft assembly. After several iterations of misjoin detection and scaffold building, a series of post-processing steps are used to fix errors and split chromosomes.
SALSA, originally published in 2017 by Ghurye, et al., is an algorithm specifically for scaffolding genome assemblies built with long reads. Another iterative scaffolder, SALSA also detects and adjusts misjoins as part of the process, outputting scaffolds of each iteration as well as a final scaffold file.
YaHS, a more recently developed tool out of the Wellcome Sanger Institute and published currently as a pre-print by Zhou et al., (2022), touts a novel method for building the contact matrices which the authors indicate may improve the assembly accuracy and contiguity, and be more robust to assembly errors.
instaGRAAL, originally published in 2014 as GRAAL by Marie-Nelly, et al., uses a Markov chain Monte Carlo (MCMC) method to find and evaluate the most likely genome given a set of genome-wide contact data through a succession of various operations such as cut, insert, flip, swap, etc.
HiCAssember, originally published in 2019 by Renschler et al., is from the same development group of HiCExplorer to take the output from that tool and then iteratively assemble the scaffolds into chromosomes.
3. Phasing
Phasing, or the separation of the maternal and paternal haplotypes inherited by an individual, often feels like the last frontier of genome sequencing. Extremely hard to do at the whole genome level, Hi-C data is helping address this need to study human disease as well as to tease apart the polyploid genomes of the most lucrative crop species.
AllHiC, originally published in 2019 by Zhang, et al., enables the chromosome-scale assembly of separate haplotypes in polyploid species. By doing a “pruning” step to remove Hi-C signals between allelic regions, the Hi-C data can be partitioned into haplotypes for downstream scaffolding.
HaploHiC uses Hi-C reads for phasing reads of unknown parental origin. Originally published in 2021 by Lindsly, et al., this tool marks Hi-C reads as haplotype-known or -unknown based on coverage of heterozygous phased SNVs/InDels.
DipAsm is an assembly tool for efficiently generating chromosome-scale, haplotype-resolved human genome assemblies. Originally published in 2020 by Garg, et al., the DipAsm method has been shown to phase >99% of heterozygous sites to 98-88% accuracy across three publicly available human genomes.
Bonus: Hi-C used directly in contig assembly
As the acknowledgment for Hi-C data for scaffolding and phasing has risen, so has the idea of efficiency and taking advantage of Hi-C data directly in the contig assembly pipeline, reducing the steps required to generate a chromosome-scale, phased genome assembly.
HiFiasm, originally published in 2021 by Cheng, et al., has been used across a wide range of species from humans to frogs to strawberries for the generation of mostly phased contig assemblies. The algorithm has recently been updated to include Hi-C data directly in the assembly pipeline, making it more effective for phasing and post-assembly scaffolding.
Summary
Hi-C data is rich in information to help build and improve genome assemblies for species across the tree of life. Whether you need a tool for publication-quality visualization of a contact map, an assembly with contigs anchored to chromosomes, or phased haplotypes to study disease or a breeding line there are many open-source tools at your fingertips. We hope this tools guide helps you efficiently meet the goals of your genome project and we look forward to including the next generation tools as developers continue to improve and innovate Hi-C data analysis.


