September 12, 2022
Share
What Makes a Genome High Quality?
It’s no surprise that the quality of a genome assembly will impact the amount and quality of insights that can be gleaned from it. So it’s worth asking the question – what makes a genome assembly “high quality”?
Here we will narrow the quality question down to four main areas that separate the draft genomes from the reference-quality genomes.
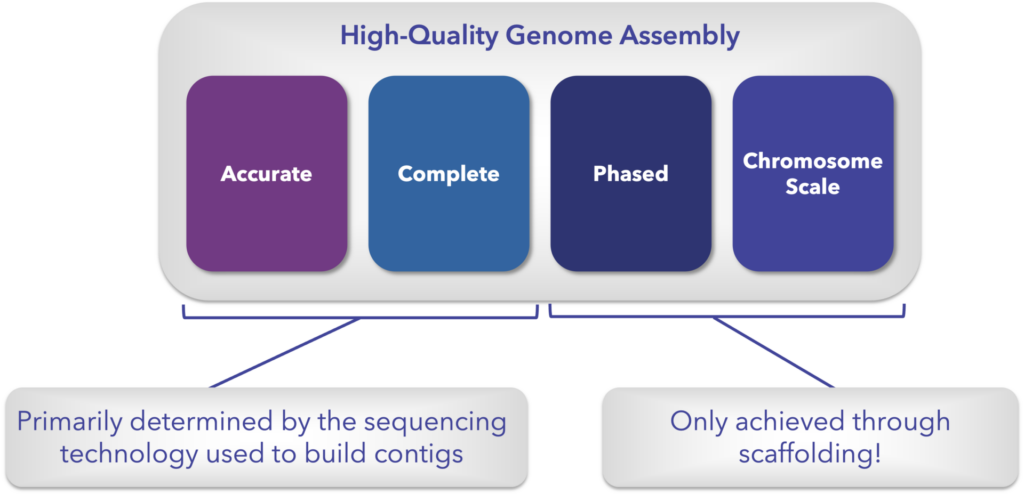
Accuracy and completeness refer to accurately representing every base in the genome and completely containing all the expressed and not expressed portions of the genome. Both of these are mostly determined at the contig level by the sequencing technology used to read and then assemble the genome into contigs.
On the other side there’s phasing – separating the sequences into their respective haplotypes, and contiguity, meaning there are no gaps in the known sequence across the entire chromosome (can also be referred to as chromosome-scale contiguity).
While DNA sequencing technologies have made big strides in achieving long, accurate contigs, they still aren’t enough to deliver chromosome-scale contiguity while also phasing haplotypes. This is no more apparent than in the recent publication of the first telomere-to-telomere human genome. They used a combination of sequencing technologies and Arima Hi-C data to finally fill all the gaps left from the original human genome project.
Four Ways Hi-C Data Improves Genome Assemblies
1. Ordering and orienting contigs
By revealing which portions of the DNA in a chromatin structure are in close proximity to each other, interaction probabilities are used to put contigs in the correct order and orientation to give a linear representation of scaffolds in a genome.
In this example, researchers used Hi-C data to place contigs from two different lentil species into the correct order and orientation and build scaffolds to represent the chromosome-scale structure of these genomes.
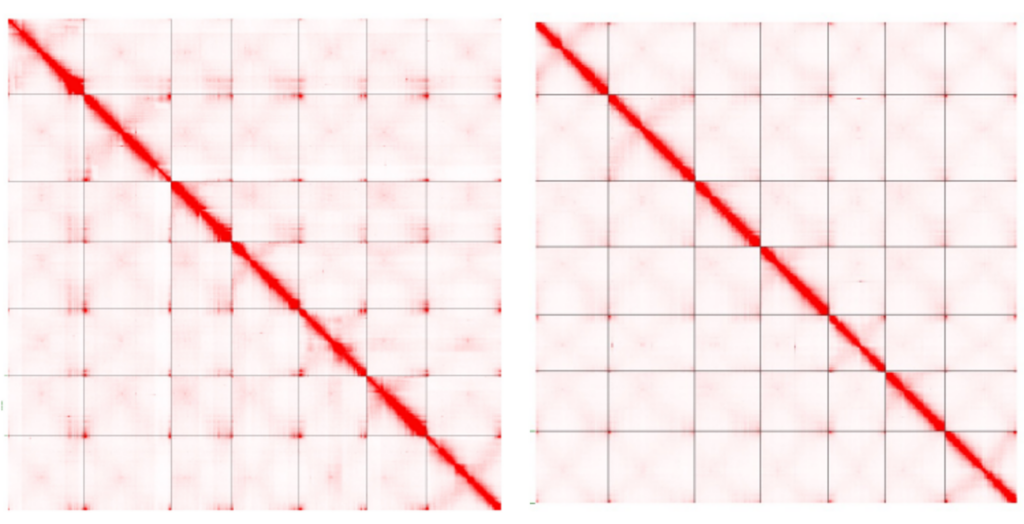
Two lentil genome assemblies with contigs scaffolded by Hi-C data from Ramsay, L., et al. (2021).
2. Fixing mis-assemblies and/or identifying structural variation
By visualizing the Hi-C data mapped to sequence data and looking for abnormal (non-linear) patterns that emerge in Hi-C contact maps, structural variation and incorrect assembling of contigs can be identified and manually corrected to improve genome assemblies.
In this example, researchers showed what incorrect assembly of contigs would look like with simulated Hi-C data, demonstrating the value in visualizing genomes with Hi-C contact maps.
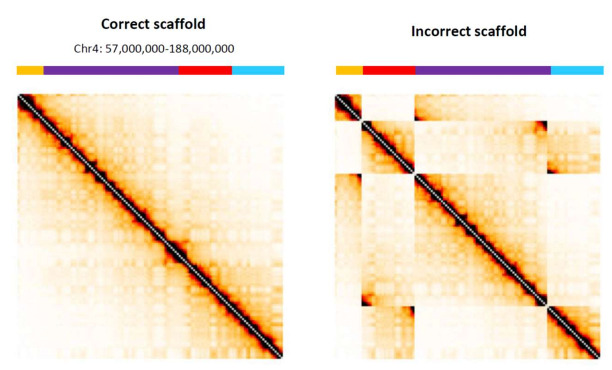
Comparison of a correctly organized Hi-C map with an incorrectly organized map from Oddes, S., et al. (2018).
3. Anchoring contigs to chromosomes
Leveraging 3D information to identify centromeric and telomeric regions, Hi-C data is used to characterize breakpoints and clearly delineate chromosomes in a genome assembly.
In this example, researchers used Hi-C data to directly link their genome sequences into the 7 pseudo-chromosomes expected for a cucumber genome.
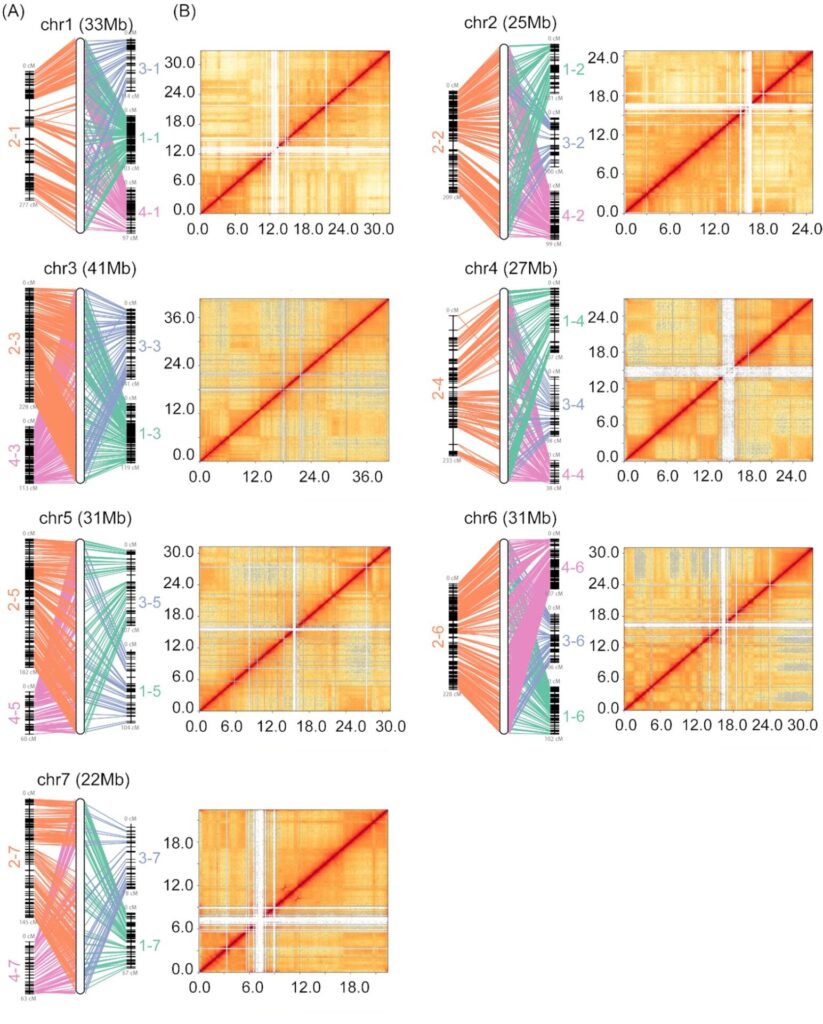
Correlation of genome assembly with genetic maps and Hi-C data for the 7 chromosomes of a cucumber genome from Li, Q., et al. (2019).
4. Phasing haplotypes
Utilizing predictable patterns of intra- vs inter-chromosomal interactions to cluster and scaffold individual haplotypes, Hi-C data helps phase contigs by clustering before scaffolding.
In this example, Hi-C data was used to identify and scaffold the four separate alleles of the wild sugarcane genome.
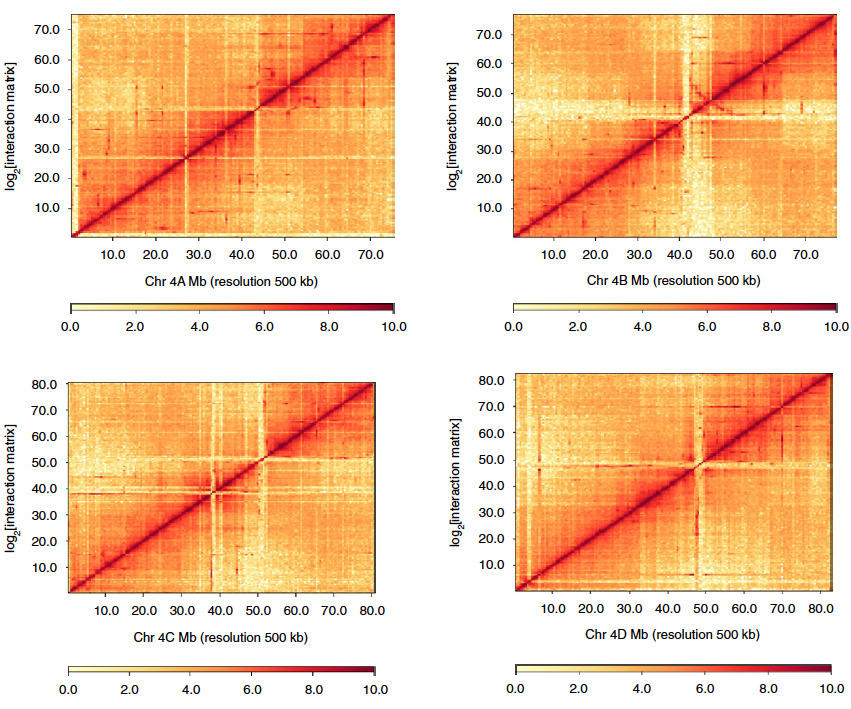
Hi-C contact maps of four homologous chromosomes in the wild sugarcane genome from Zhang, X., et al. (2019).
A High-Quality Genome Assembly is a Strong Foundation
To truly understand genome biology, complete genomes are needed. Complete genomes are ones that are accurate, haplotype-resolved, and chromosome-scale. It’s possible that DNA sequencing technologies might provide all of the requirements for a reference genome in the future, but for now Hi-C data helps close the gap between contigs and a high-quality genome assembly, enabling groundbreaking discoveries in diverse fields of science.

Learn more about adding Arima Hi-C to your genome assembly research with our kits or services.
Resources
Ramsay, L., et al. (2021). Genomic rearrangements have consequences for introgression breeding as revealed by genome assemblies of wild and cultivated lentil species. BioRxiv, 2021.07.23.453237.
Oddes, S., et al. (2018). Three invariant Hi-C interaction patterns: Applications to genome assembly. BioRxiv.
Li, Q., et al. (2019). A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience, 8(6), giz072.
Zhang, X., et al. (2019). Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nature Plants, 5(8), 833–845.


