December 21, 2021
Share
Seventeen years ago, the Human Genome Project was completed through painstaking manual effort and tremendous cost, heralding the beginning of the genomics era. We are now on the cusp of yet another genomics revolution. By 2031, global initiatives, such as the Earth BioGenome Project, will generate an unprecedented repository of high-quality reference genome assemblies for every known eukaryotic species – all 1.5 million of them.
Genomic data is used by diverse biological fields, from conservation to biomedical research, and the quality of this genetic information is paramount. The Earth BioGenome Project is working towards the ultimate goal of sequencing all complex life on Earth and relies on affiliated project networks, including the Vertebrate Genome Project (VGP) and the Darwin Tree of Life, to generate these high-quality, annotated reference genomes.

Arima Genomics has partnered with various initiatives, including the VGP and the Darwin Tree of Life, to harness our Hi-C technology to improve the contiguity, quality, and phasing of the assemblies.
A major challenge for conducting high-quality genomic research is that most current genome assemblies are riddled with errors. Parts of genes are missing, some are incorrectly assembled, while others are completely missing from the assemblies despite pieces found in the raw sequence reads. Technological advances, improved computational methods, and the ever-decreasing cost of sequencing enabled the Vertebrate Genomes Project to pursue the ambitious goal of producing a reference genome assembly for each of the 71,657 extant vertebrate species on Earth. Their first step was to test and improve genome sequencing and assembly approaches toward the goal of creating high-quality, near-error free and gapless, haplotype phased and annotated reference genomes.
The Darwin Tree of Life program similarly aims to sequence the genomes of 70,000 species of eukaryotic organisms in Britain and Ireland with the goal towards transforming our approach to biology, conservation, and biotechnology. Wellcome Open Research releases each Tree of Life genome assembly as a micropublication called a ‘Genome Note’, which summarizes the origin of the specimen used for sequencing, the methods used to extract and sequence the genetic material, and methods used to assemble and polish the assemblies.
Arima Genomics has partnered with various initiatives, including the VGP and the Darwin Tree of Life, to harness our Hi-C technology to improve the contiguity, quality, and phasing of the assemblies. This approach provides more comprehensive information than genomic sequencing alone. Hi-C technology is used specifically to detect and correct errors in assemblies – for example, false duplications – and orient sequences to chromosomes. Hi-C analysis facilitates complete haplotype phasing and resolution of long repeat regions (telomeres, centromeres, sex chromosomes).
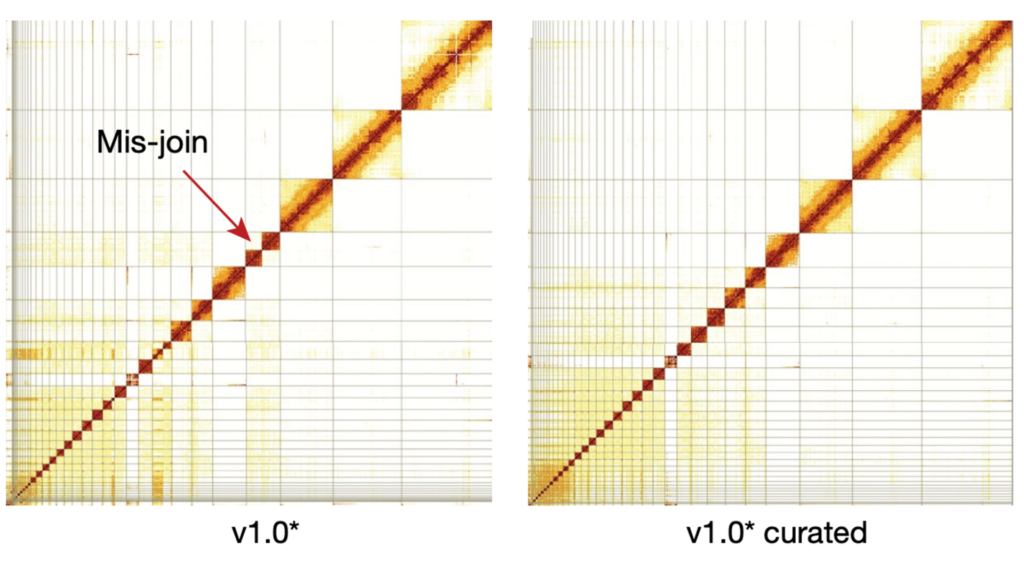
Comparative analysis of Anna’s hummingbird genome assemblies to optimize sequencing and assembly strategy for high-quality reference genomes. Hi-C interaction heat maps shown before and after manual curation; red arrow indicates a mis-join that was corrected during curation.
The high-quality reference genomes generated by the Vertebrate Genome Project have contributed to multiple scientific disciplines. Arima Genomics is proud to have facilitated these efforts, a few of which are highlighted here:
Marmoset Genome Reveals Biomedical Insights

Marmoset, courtesy of Leszek Leszczynski, CC BY 2.0, via Wikimedia Commons.
Conservation Genomics to Save the Vaquita

Vaquita courtesy of Alfokrads, CC BY-SA 4.0, via Wikimedia Commons
Biology and Evolution of Bat Adaptations

Velvety free-tailed bat (Molossus molossus) courtesy of Cardoso Cláudio V, et al. (2020) Zoologia, via Creative Commons
Explore the Vertebrate Genomes Project collection page for additional VGP publications and information.
The Genome Notes, published as part of the Wellcome Open Research Tree of Life Gateway, are intended to promote the discovery and use of the sequence datasets by providing a detailed description of each. All submitted Genome Notes can be found on the Tree of Life Gateway; a select few recently published genomes are listed below:
- Devil’s coach horse (Ocypus olens)
- Small copper or common copper (Lycaena phlaeas)
- Common toad (Bufo bufo)
- Common frog (Rana temporaria)
- Tapered dronefly (Eristalis pertinax)
- Green-veined white butterfly (Pieris napi)
- Predatory ribbon worm (Lineus longissimus)
- Garden bumblebee (Bombus hortorum)
- Peach blossom moth (Thyatira batis)
- Glanville fritillary (Melitaea cinxia)
Discover the more than 100 published assemblies generated with Arima Hi-C technology on NCBI, learn more about adding Hi-C to your genome assembly research or request a project consultation.
Resources
Rhie A, et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021). https://doi.org/10.1038/s41586-021-03451-0
Yang C, et al. Evolutionary and biomedical insights from a marmoset diploid genome assembly. Nature 594, 227–233 (2021). https://doi.org/10.1038/s41586-021-03535-x
Jebb D, et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature 583, 578–584 (2020). https://doi.org/10.1038/s41586-020-2486-3
Morin, PA et al. Reference genome and demographic history of the most endangered marine mammal, the vaquita. Molecular Ecology Resources. 21: 1008-1020 (2021). https://doi.org/10.1111/1755-0998.13284


