October 19, 2022
Share
To enable the generation of a complete view of human genetic diversity, the Human Pangenome Reference Consortium was formed with the goal of creating high-quality, cost-effective diploid genome assemblies for a pangenome reference.

In their paper Semi-automated assembly of high-quality diploid human reference genomes published today in Nature, an international team of scientists and bioinformaticians have published their work to determine which combination of current genome sequencing and assembly approaches yields the most complete and accurate diploid genome assembly with minimal manual curation.
In this extensive study, termed an assembly bakeoff or “assemblathon” the teams employed various sequencing technologies, including ONT and PacBio long reads and many assembly pipelines, then paired that with optical mapping and Hi-C linked reads for scaffolding. Using their initial findings, they then developed a pipeline that combined the best practices of all approaches and used it to generate a higher-quality diploid de novo assembly.
Using Hi-C to Scaffold and Phase High-Quality Genomes – Highlights
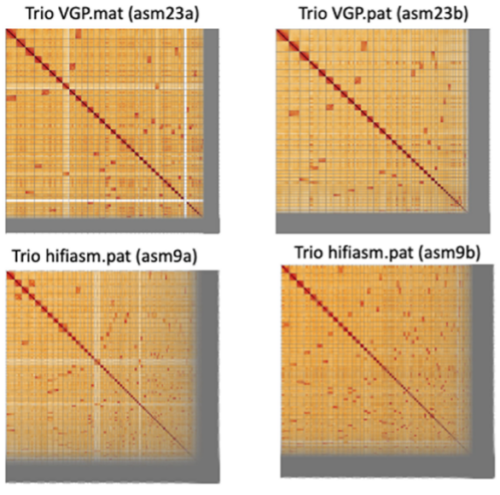
Hi-C contact maps for showing maternal (mat) and paternal (pat) haplotype assemblies using the trio VGP scaffolded assembly (asm23) and trio hifiasm contig only assembly (asm9) approaches.
- As part of the bake-off of approaches to generate highly contiguous phased assemblies, the team used Hi-C interaction plots to identify up to several hundred scaffolding errors per assembly. These errors included missed joins, misjoins, and erroneous inversions or false duplications classified as other errors.
- From the information gathered from the extensive comparisons of 23 assembly approaches, the authors report that methods that used highly accurate long reads and parent-child data with graph-based haplotype phasing using Hi-C data during the assembly process outperformed those that did not. Below is the pipeline the group identified as optimal for those seeking to obtain the highest-quality assemblies and contribute to the human pangenome.
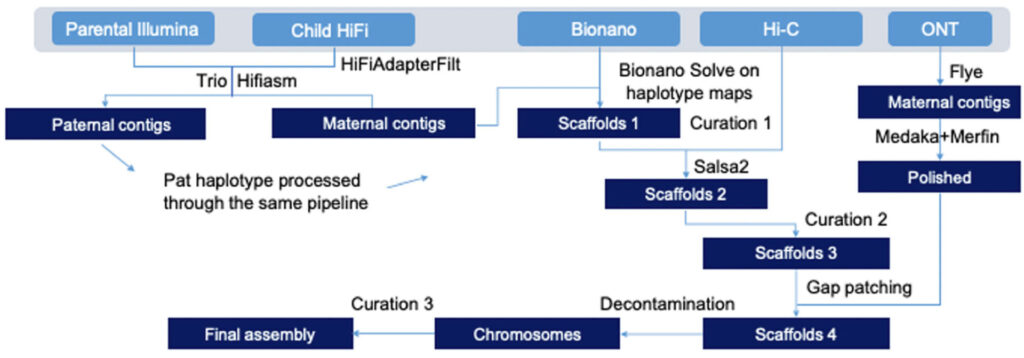
Trio pipeline used for pangenome alignment and generation of high-quality HPRC-HG002 v1.0 diploid assemblies.
- The final diploid assembly they produced using the Trio HPRC v1.0 pipeline, and Trio hifiasm for scaffolding yielded a high-quality genome with assembly metrics on par with the first complete human genome assembly (T2T-CHM13 v1.1), including near-complete haplotype separation of scaffolds as seen in the Hi-C profiles.
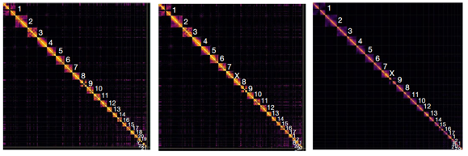
Hi-C contact maps to the final curated HPRC-HG002 paternal (left) and maternal (middle) assemblies in comparison to the CHM13 assembly (right) with chromosome numbers.
Although the team had substantial success with generating near-complete phased haplotypes using the trio approach, future efforts will be necessary to develop a phasing method that does not require parental sequence data to produce diploid reference assemblies for human and non-human organisms where parental data is unavailable. The authors note that Hi-C data for haplotype phasing is a promising alternative, as Hi-C data contains within-chromosome haplotype information for an individual.
Taken together, the data presented in this study demonstrate that integration of both HiFi and ONT-UL data in a diploid assembly graph, combined with long-range phasing information from Hi-C or Strand-seq could soon enable automated T2T diploid genome assemblies. The findings from this work serve as a foundation for assembling near-complete diploid human genomes at scale to fuel the production of the human reference pangenome.
Learn more about Arima Hi-C for genome assembly and our products and services.


