March 28, 2023
Share
Characterizing cancer genomes at the chromosome-scale with high resolution is critical for advancing personalized diagnosis and disease management. However, current short-read sequencing and analytical tools are not designed for cancer genomes and are insufficient for chromosome-scale analysis of structural variant (SV) landscapes. Recent work by Shilpa Garg at the Technical University of Denmark and the University of Copenhagen combined high-resolution sequencing techniques and a novel computational platform to accurately and precisely reconstruct cancer genomes.
Garg applied long and accurate PacBio HiFi sequencing and long-range Arima Hi-C data to melanoma COLO829 cancer cell lines to examine cancerous mutations at base and haplotype resolution. These data were then processed through a novel, high throughput graph-based computational tool, pstools. Garg demonstrates that combining these approaches enables precise characterization of SV landscapes of cancer genomes, outperforming many existing methods.
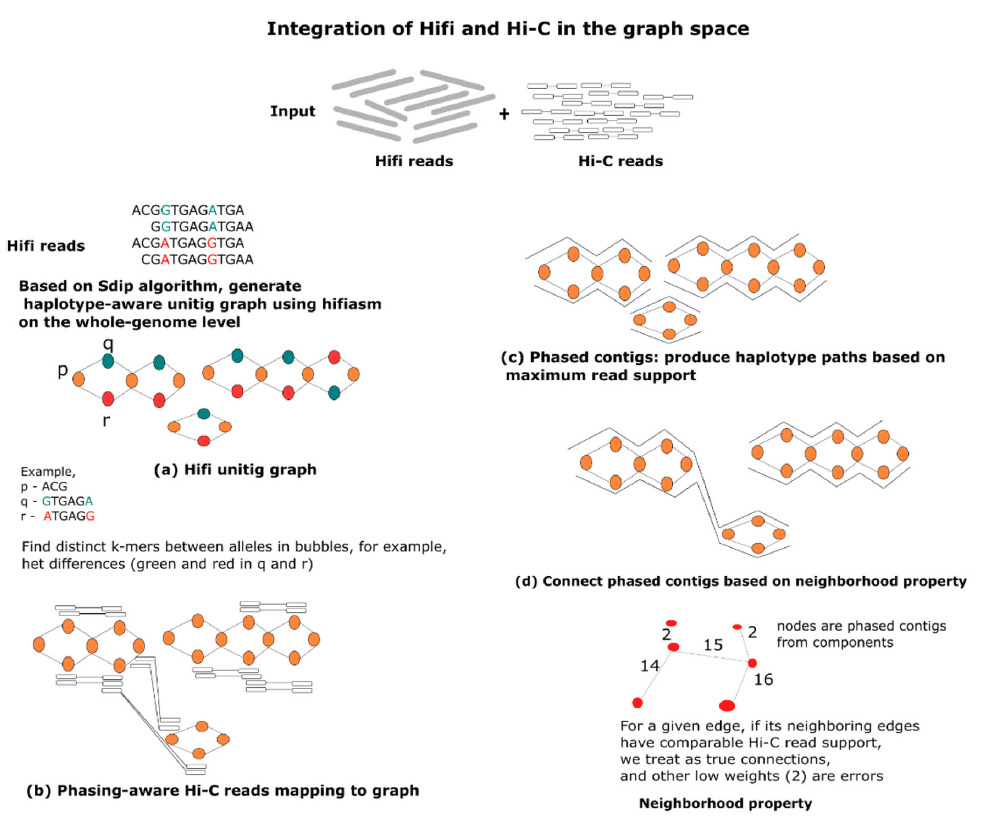
Workflow for processing Hifi and Hi-C reads with pstools, a graph-based algorithm for chromosome-scale genome analyses.
With the pstools approach, the author delineates several characteristics of the cancer genome that are often missed with current analytical tools such as HiCanu, Flye, trio-hifiasm, and salsa2. Importantly, pstools is fast and accurate, enabling high-throughput and routine analyses of fully phased sequences at the chromosome scale.
Shilpa Garg leveraged 3D genomic data generated using the Arima genome-wide HiC kit to obtain Hi-C reads that were then analyzed using pstools. Published in Nature Communications, this workflow provides a foundation for streamlining cancer genome research, with important implications for improving personalized cancer diagnosis and treatment.
3D Genomics Insights Help Enable Chromosome-scale Haplotype-resolved Reconstruction in Cancer Genomics – Highlights
- Combining HiFi, Hi-C, and graph-based computational approaches in COLO829 cancer cell lines facilitated the precise identification and characterization of somatic SV calls in critical repeat elements.
- With an integrative graph algorithm, this protocol captures, at chromosome-scale, inter- and intra-chromosomal structural genome complexity, haplotype information, produces complete phased genomes, and can perform polyploid phasing in the presence of aneuploidy and transposable elements.
- Benchmarking this high-resolution sequencing and graph-based approach on healthy human samples revealed that some existing analysis methods cannot generate assembly sizes appropriate for cancer genomics. However, the graph-based approach is capable of larger assemblies for cancer genomics and boasts faster runtimes (12 hours) and comparable quality to trio-hifiasm+salsa2 methods.
- Moreover, compared to trio-hifiasm+salsa2, pstools generates higher quality phased scaffolds without trio information.
- Pstools is also capable of finding precise and accurate chromosome-level SV landscapes. With it, the researchers identified 19,956 insertions,14,846 deletions, 421 duplications, 52 inversions, and 498 translocations. Additionally, pstools outperformed several other methods in SV callsets, including detecting somatic SV calls in repeat elements that were missed in other approaches, demonstrating a major advantage of pstools over existing technologies.
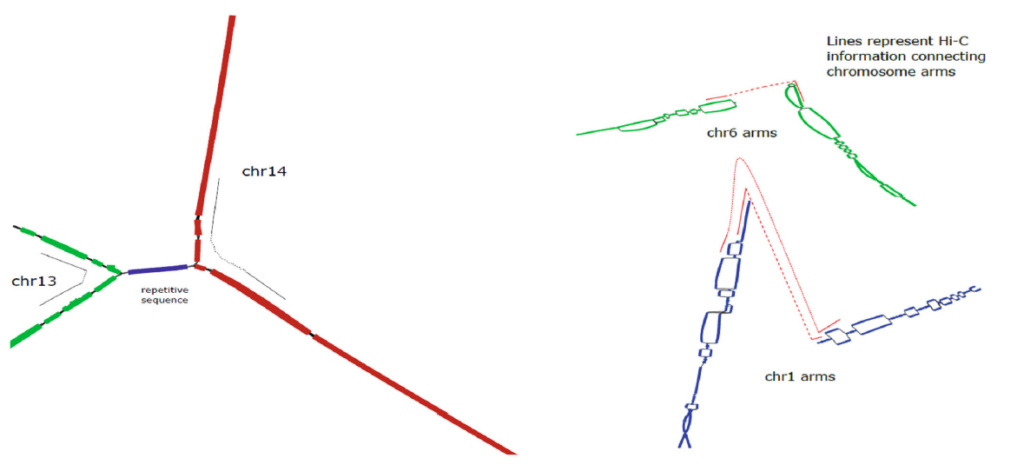
When incorporated into the pstools algorithm, Arima Hi-C data facilitates disentangling the chromosomes (left) and accurately connecting the starting regions of chromosome arms (right).
In the Nature Communications article, Garg details novel sequencing and computational protocols for comprehensive cancer genome reconstruction. While highly promising, the author acknowledges that pstools is limited in its ability to characterize somatic genetic variation at low variant allele frequencies, requiring future studies that explore single-cell, long=read approaches.
Learn more about cancer genomics or request a project consultation.
Resources
Garg, S. (2023). Towards routine chromosome-scale haplotype-resolved reconstruction in cancer genomics. Nature Communications, 14(1), 1358.


